Vitamins are important helpers for your body. There are two main kinds:
- Fat soluble vitamins – stay in your body for a long time – A, D, E, and K
- Water soluble vitamins – don’t stay in your body for very long
1. Fat-soluble vitamins
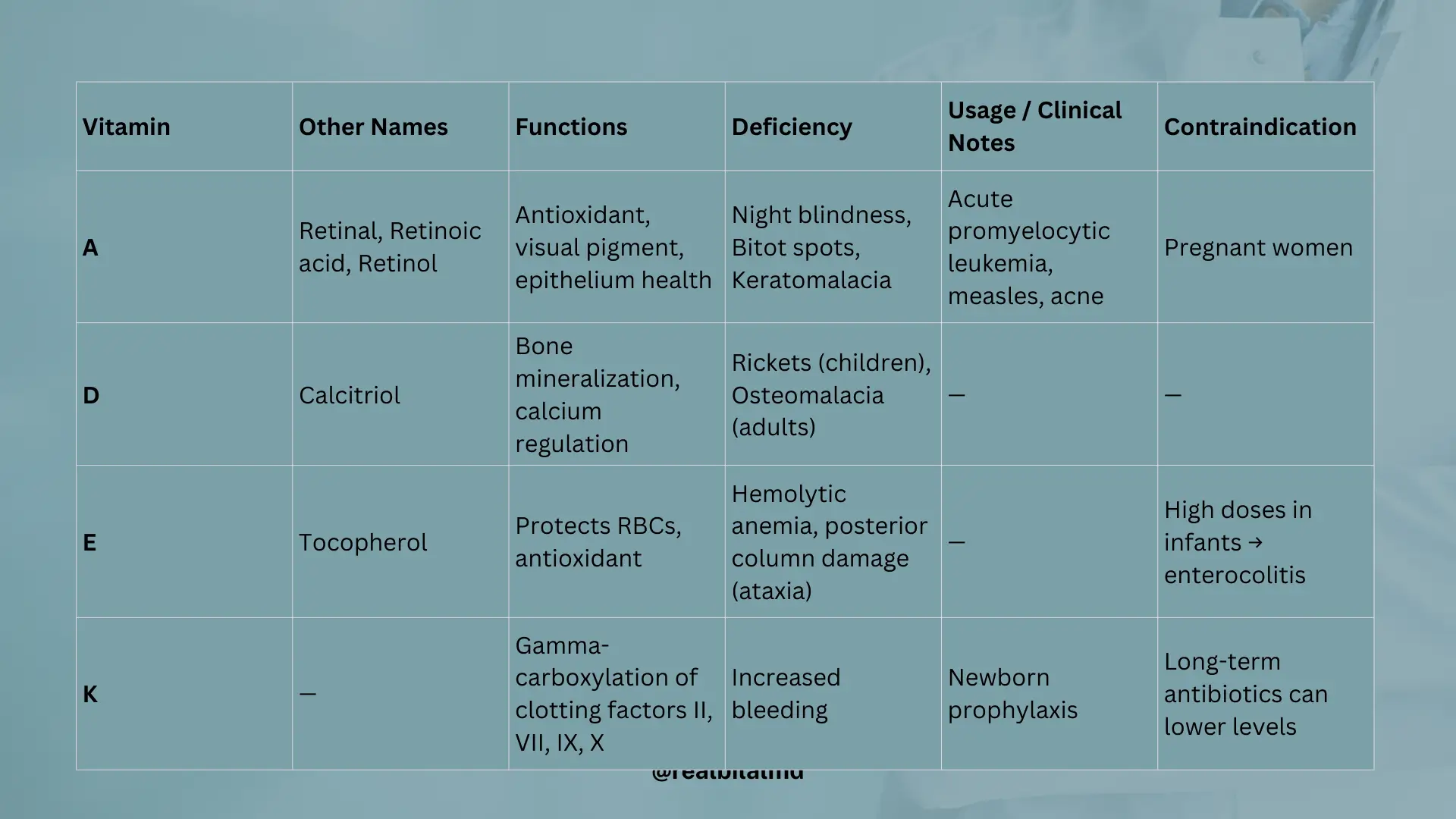
1. Vitamin A
Vitamin A has several names, including retinal, Retnoic acid and Retinol.
What it does:
- It’s like a superhero that fights off bad stuff in your body (antioxidant).
- It helps you see, especially in low light conditions (visual pigment).
- It keeps your skin and other body coverings healthy (essential for epithelium).
Usage:
- To help with a type of blood cancer called acute promyelocytic leukemia.
- When kids have measles.
- For skin problems like acne.
Deficiency:
- You might have trouble seeing at night (nyctalopia).
- You can get dry spots on your eyes (bitot spots on conjunctiva).
- Your eyes might get sick (keratomalacia that lead to degeneration of cornea).
Contraindication:
Pregnant women should not take Vitamin A.
2. Vitamin D
Vitamin D is also called Calcitriol.
Source
Sunlight is a great source!
Storage
Vitamin D is stored in liver in form of 25-hydroxyvitamin D (25(OH)D)
Activation
- Parathyroid hormone (PTH) tells the fto turn on a special enzyme called 1-alpha-hydroxylase.
- This enzyme changes vitamin D into its active form — 1,25-dihydroxyvitamin D3 — in a part of the kidney called the proximal convoluted tubule (PCT).
If
- Normal vitamin D helps build strong bones by adding more calcium to them (this is called mineralization).
- Too much vitamin D can actually do the opposite — it tells the body to take calcium out of the bones, which can make them weaker (this is called resorption)
Deficiency
- In children, soft and bendy bones can cause bowed legs — this condition is called Rickets. It can also happen due to a condition called renal (kidney) dystrophy.
- In adults, weak bones cause bone pain and muscle weakness — this is called Osteomalacia.
In both cases:
- Alkaline phosphatase (ALP) goes up.
- Parathyroid hormone (PTH) goes up,
- Calcium (Ca²⁺) may go down,
- Phosphate (PO₄³⁻) goes down,
3. Vitamin E
Vitamin E is also called Tocopherol.
Function
Protects red blood cells from oxidative stress.
Deficiency
- Low vitamin E can make red blood cells break too easily — this causes hemolytic anemia.
- It can also damage the nerves, especially in the spine (posterior columns), leading to balance problems (ataxia).
- Too much vitamin E in infants may cause enterocolitis (gut inflammation).
- Lack of vitamin E and B12 can both cause neurological problems.

4. Vitamin K
Source:
- Found in green leafy vegetables
- Also made by bacteria in the intestine
Activation:
- In the liver, an enzyme called epoxide reductase activates vitamin K
- Vitamin K helps with a process called gamma-carboxylation of glutamic acid, which adds a carboxyl group to clotting factors
Function:
Needed to activate clotting factors II, VII, IX, and X (2, 7, 9, 10)
Vitamin K – Deficiency
- Causes increased bleeding because blood can’t clot properly
Diagnosis:
- Bleeding time (BT): Normal (platelets are fine)
- Prothrombin time (PT): Prolonged – reflects the extrinsic pathway (factor VII)
- aPTT (activated partial thromboplastin time): Prolonged – reflects intrinsic pathway (factors IX, X)
Extra Info:
- Newborns are given Vitamin K to prevent hemorrhagic disease of the newborn
- Long-term antibiotic use can lower Vitamin K by killing gut bacteria that make it
Here are other materials for NLE NRE step 1
2. Water-Soluble Vitamins
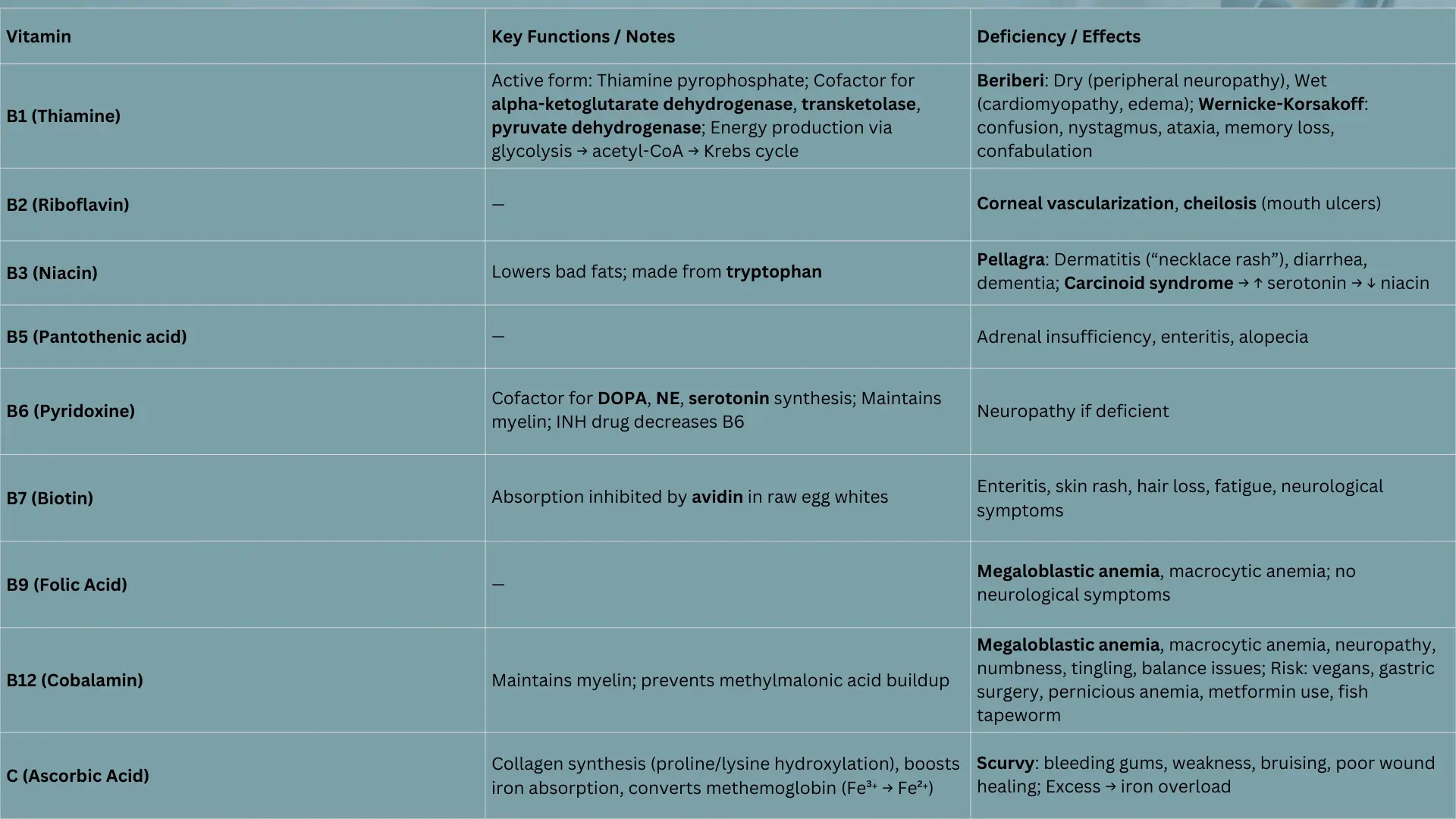
1. Vitamin B1 (Thiamine)
It is found in the outer part of rice. Thiamine is a inactive form and thiamine pyrophosphate is a active form.
Function
Enzymes That Need Vitamin B1 (Thiamine) to Work:
- Alpha-ketoglutarate dehydrogenase – in the Krebs cycle
- Transketolase – in the HMP shunt (Pentose Phosphate Pathway)
- Pyruvate dehydrogenase – the link between glycolysis and Krebs cycle
All of these enzymes need B1 as a cofactor to help produce energy.
- Glucose (6 carbon) is broken down into pyruvic acid (3 carbon) through glycolysis.
- Pyruvic acid is then converted to acetyl-CoA (2 carbon) in the linker step.
- Acetyl-CoA enters the Krebs cycle, where it helps produce ATP (energy).
Deficiency
Beriberi – Caused by Vitamin B1 (Thiamine) Deficiency
- Dry Beriberi:
Affects nerves and muscles, causing muscle weakness and numbness (peripheral neuropathy). - Wet Beriberi:
Affects the heart, leading to dilated cardiomyopathy, fast heart rate, and fluid buildup (swelling).
Wernicke-Korsakoff Syndrome – Caused by Vitamin B1 (Thiamine) deficiency
- It can lead to brain damage and serious neurological symptoms.
Wernicke’s Encephalopathy (early stage):
- Confusion
- Strange eye movements (nystagmus) and eye muscle weakness (ophthalmoplegia)
- Trouble walking (ataxia)
Often due to poor nutrition
Korsakoff Syndrome (late stage):
- Memory loss and confabulation (making up stories)
Often caused by chronic alcohol use, which reduces vitamin absorption
2. Vitamin B2 (Riboflavin)
Deficiency
- Corneal vascularization
- cheilosis – ulcers in mouth
3. Vitamin B3 (Niacin, Nicotinic acid)
It can help lower bad fats in your blood. Your body can make it from a special building block called Tryptophan.
Deficiency
You can get a sickness called Pellagra.
- This causes:
- Neckless rashes.
- Diarrhea (tummy troubles).
- Dementia (trouble thinking clearly).
- It can also cause gut problems and hair loss.
Carcinoid Syndrome
- In this condition, serotonin levels increase because certain tumors (carcinoid tumors) make too much of it.
- Tryptophan, the amino acid used to make serotonin, gets used up.
- This means less tryptophan is available to make niacin (Vitamin B3) → leading to niacin deficiency.
So in carcinoid syndrome:
- ↑ Serotonin
- ↓ Niacin → can lead to pellagra (diarrhea, dermatitis, dementia)
4. Vitamin B5
Deficiency
- Adrenal gland insufficiency
- Enteritis
- Alopecia
5. Vitamin B6 (Pyridoxine)
- What it does: It helps your body make important brain chemicals like Dopamine and Serotonin. It also helps keep the protective covering around your nerves healthy.
- Important note: A medicine called INH can stop Vitamin B6 from working.
6. Vitamin B5
Deficiency
- Adrenal gland insuficiency
- Enteritis
- Alopecia
7. Vitamin B5 (Pyridoxine)
Usage
Used as a cofactor in the synthesis of important neurotransmitters:
- Dopamine (DOPA)
- Norepinephrine (NE)
- Serotonin
Function
Helps maintain the myelin sheath, which protects nerve fibers.
The drug Isoniazid (used for tuberculosis) can decrease Vitamin B6 (Pyridoxine) levels, leading to neuropathy if not supplemented.
8. Vitamin B7 (Biotin)
Raw egg whites contain a protein called avidin, which binds to biotin and prevents its absorption.
Eating too many raw egg whites can lead to biotin deficiency.

Effects of Biotin Deficiency:
- May increase risk of enteritis (inflammation of the intestines)
- Skin rash
- Hair loss
- Fatigue
- Neurological symptoms
9. Vitamin B9 (Folic Acid)
Deficiency
- No neurological symptoms (unlike Vitamin B12 deficiency)
- Causes megaloblastic anemia
- Also leads to macrocytic anemia (large red blood cells)
10. Vitamin B12 (Cobalamin)
Deficiency
- Causes megaloblastic anemia
- Causes macrocytic anemia (large red blood cells)
- Yes — neurological symptoms are present
- Numbness, tingling, balance problems
Why neurological symptoms occur:
- Methylmalonic acid increases in B12 deficiency
- These harm the myelin sheath, damaging nerves
- This leads to buildup of toxic compounds (like abnormal succinyl-CoA)
Who Is at Risk for Vitamin B12 (Cobalamin) Deficiency?
- Leads to macrocytic (megaloblastic) anemia and neurological symptoms
- Strict vegetarians (vegans) – B12 is found mainly in animal products
- People with stomach surgery (e.g., gastric bypass) – reduced intrinsic factor
- Pernicious anemia – autoimmune condition that reduces B12 absorption
- Metformin users – long-term use can interfere with B12 absorption
- Infection with Diphyllobothrium latum – a fish tapeworm that absorbs B12 from the gut
11. Vitamin C (Ascorbic Acid)
Functions:
- Helps in collagen synthesis by aiding the hydroxylation of proline and lysine
- Boosts iron absorption from the intestine by keeping iron in its ferrous (Fe²⁺) form
Risk:
Excess vitamin C can increase iron absorption too much, leading to iron overload (hemochromatosis).
Vitamin C Deficiency
- Causes Scurvy
- Symptoms include bleeding gums, weakness, bruising, and poor wound healing
- Due to impaired collagen synthesis
Other Functions of Vitamin C
- Converts methemoglobin (Fe³⁺) back to normal hemoglobin (Fe²⁺)
- Helps oxygen bind properly to red blood cells
3. Protein Energy Malnutrition
1. Kawashiorkor
- Caused by protein deficiency, especially low albumin
- Albumin maintains oncotic pressure in blood vessels
- Low albumin → decreased oncotic pressure → fluid leaks out → edema
Remember the Mnemonic: MEALS
- M – Malnutrition (mainly protein)
- E – Edema (especially in legs and face)
- A – Anemia
- L – Liver (fatty) → due to impaired lipoprotein synthesis
- S – Skin lesions (peeling, flaky paint appearance)
2. Marasmus
Causes
- Caused by calorie deficiency (not enough total energy intake)
- Protein intake may be normal, but overall energy is too low
- The body starts using fat and muscle for energy
- Leads to severe muscle wasting and weight loss
- No edema (unlike Kwashiorkor)
Key Features:
- Often affects infants and young children
- Severe weight loss
- Muscle wasting (thin, lean appearance)
- No fat stores
- Alert but weak child

1. Collagen Protein (Most Abundant)
Collagen is the most common protein in your body. Different types of collagen are found in different parts of the body:
- Type I – Found in bones, teeth, tendons, and skin
- Type II – Found in cartilage (important for joints)
- Type III – Found in healing tissues, skin, and blood vessels (also in fetal tissue)
- Type IV – Found in basement membranes (a thin layer supporting tissues, like in kidneys)
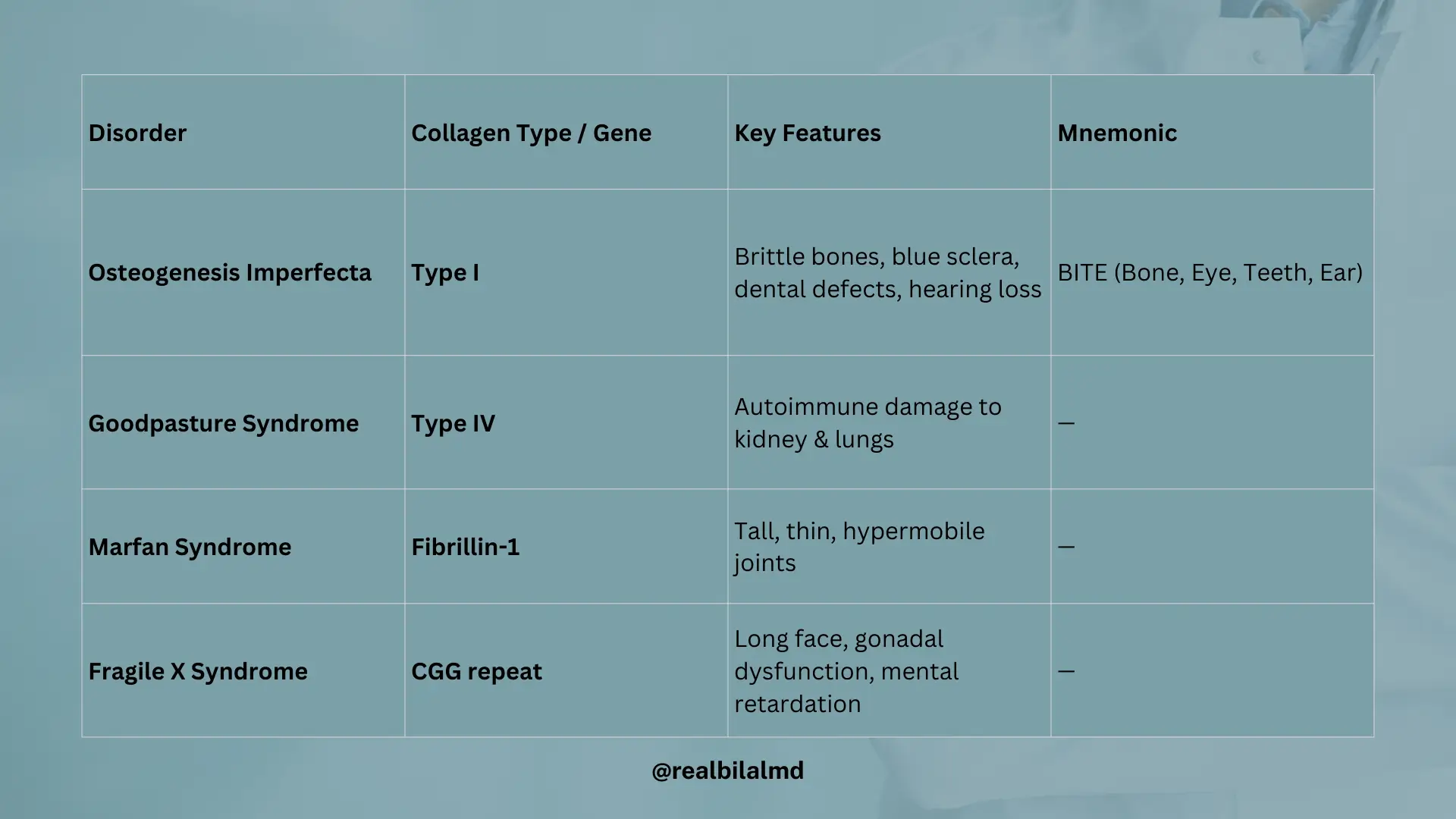
1. Osteogenesis Imperfecta (OI)
- A genetic disorder caused by decreased production of Type I collagen
- Leads to weak bones that fracture easily, even with minor injury
- Often seen in infants and children
Key Features:
- Brittle bones → frequent fractures
- Blue sclera → due to thin connective tissue in the eyes
- Tooth imperfections → weak dentin (dentinogenesis imperfecta)
- Hearing loss → due to abnormal middle ear bones
Mnemonic: BITE
- B – Bone problems (fractures)
- I – Eye: Blue sclera
- T – Teeth defects
- E – Ear issues (hearing loss)
2. Goodpasture Syndrome
- An autoimmune disease where the body makes antibodies against Type IV collagen
- Type IV collagen is found in the basement membrane of the lungs (alveoli) and kidneys (glomeruli)
Effects:
- Kidney damage → glomerulonephritis → blood in urine (hematuria), kidney failure
- Lung damage → bleeding in alveoli → coughing up blood (hemoptysis)
2. Rate Limiting Enzymes (Body Helpers)
These enzymes act like “special workers” that control the speed of important body processes:
| Pathway | Key Regulatory Enzyme |
|---|---|
| Glycolysis | Phosphofructokinase-1 (PFK-1) |
| Krebs Cycle | Isocitrate Dehydrogenase |
| Cholesterol Synthesis | HMG-CoA Reductase |
| Ketone Body Synthesis | HMG-CoA Synthase |
| Glycogen Synthesis | Glycogen Synthase |
| Glycogen Breakdown (Glycogenolysis) | Glycogen Phosphorylase |
| Urea Cycle | Carbamoyl Phosphate Synthetase I (CPS-I) |
3. Glycogen Storage Diseases
These are problems where your body can’t properly store or use glycogen (stored sugar).
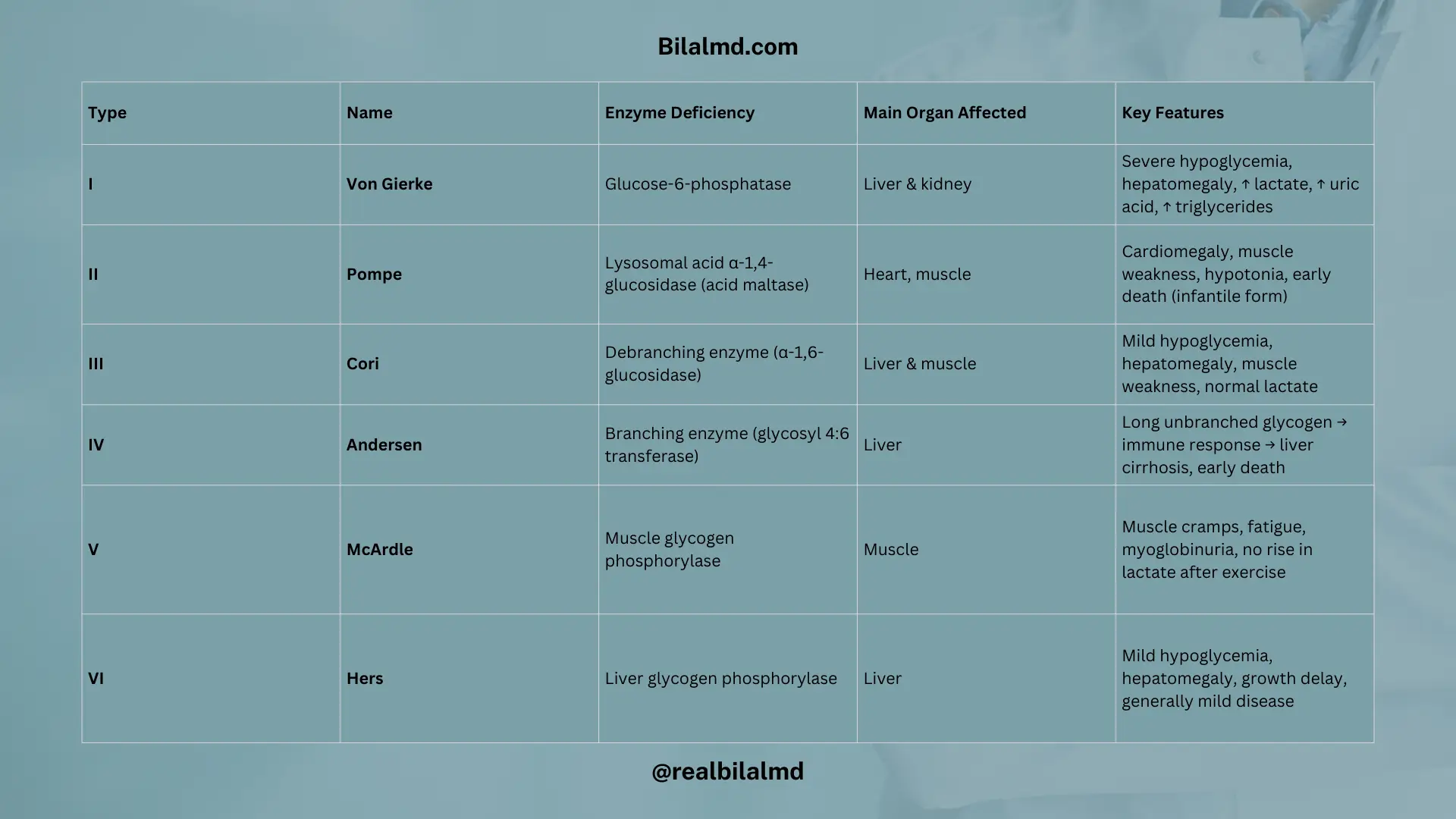
1. Von Gierke Disease (Glycogen Storage Disease Type I)
- Caused by Glucose-6-phosphatase deficiency
- Your body can’t break down glycogen properly to release glucose
- This leads to severe fasting hypoglycemia
Key Features:
- Severe hypoglycemia (especially during fasting)
- Hepatomegaly (enlarged liver due to glycogen buildup)
- High uric acid → risk of gout
- High triglycerides → lipid problems
- Lactic acidosis → due to impaired glucose metabolism
2. Pompe Disease (Glycogen Storage Disease Type II)
- Caused by a deficiency of acid maltase
(also called lysosomal α-1,4-glucosidase) - This enzyme is needed to break down glycogen inside lysosomes
Key Features:
- Massive glycogen buildup in the heart, muscles, and liver
- Cardiomegaly (enlarged heart)
- Muscle weakness (hypotonia – “floppy baby”)
- Respiratory problems
- Early death in infantile form if untreated
3. Cori Disease (Glycogen Storage Disease Type III)
- Caused by a deficiency in the debranching enzyme (also called α-1,6-glucosidase)
- The body can’t fully break down glycogen, so abnormal glycogen with short outer branches builds up in the liver and muscles
Key Features:
- Mild hypoglycemia (less severe than Von Gierke)
- Hepatomegaly (enlarged liver)
- Muscle weakness in some cases
- Normal blood lactate levels (unlike Von Gierke)
- Can cause growth delay in children
4. Andersen Disease (Glycogen Storage Disease Type IV)
- Caused by a defect in the branching enzyme (also called glycosyl-4:6-transferase)
- This enzyme normally adds branches to glycogen chains, making them soluble and compact
What Happens:
- Without branching, glycogen forms abnormally long chains
- These look foreign to the body and trigger an immune response
- Leads to liver damage and cirrhosis
Key Features:
- Liver failure (often fatal in early childhood if untreated)
- Failure to thrive in infancy
- Hepatomegaly (enlarged liver)
- Progressive liver cirrhosis
5. McArdle Disease (Glycogen Storage Disease Type V)
- Defect: Muscle glycogen phosphorylase
- Problem: Muscles can’t access glycogen for energy during exercise
Key Features:
- “Second wind” phenomenon (symptoms improve after rest)
- Muscle cramps and fatigue during exercise
- No rise in blood lactate after exercise
- Myoglobinuria (dark urine after exercise)
Key Features:
- Massive glycogen buildup in the heart, muscles, and liver
- Cardiomegaly (enlarged heart)
- Muscle weakness (hypotonia – “floppy baby”)
- Respiratory problems
- Early death in infantile form if untreated
6. Hers Disease (Glycogen Storage Disease Type VI)
- Defect: Liver glycogen phosphorylase
- Problem: Liver can’t break down glycogen to release glucose
Key Features:
- Mild fasting hypoglycemia
- Hepatomegaly
- Growth delay in children
- Usually milder than Von Gierke or Cori disease
| Type | Name | Enzyme Deficiency | Main Organ Affected | Key Features |
|---|---|---|---|---|
| I | Von Gierke | Glucose-6-phosphatase | Liver & kidney | Severe hypoglycemia, hepatomegaly, ↑ lactate, ↑ uric acid, ↑ triglycerides |
| II | Pompe | Lysosomal acid α-1,4-glucosidase (acid maltase) | Heart, muscle | Cardiomegaly, muscle weakness, hypotonia, early death (infantile form) |
| III | Cori | Debranching enzyme (α-1,6-glucosidase) | Liver & muscle | Mild hypoglycemia, hepatomegaly, muscle weakness, normal lactate |
| IV | Andersen | Branching enzyme (glycosyl 4:6 transferase) | Liver | Long unbranched glycogen → immune response → liver cirrhosis, early death |
| V | McArdle | Muscle glycogen phosphorylase | Muscle | Muscle cramps, fatigue, myoglobinuria, no rise in lactate after exercise |
| VI | Hers | Liver glycogen phosphorylase | Liver | Mild hypoglycemia, hepatomegaly, growth delay, generally mild disease |
4. Muscle Diseases (Muscular Dystrophies)
1. Duchenne Muscular Dystrophy (DMD)
Genetics:
- X-linked recessive disorder
- Affected boys inherit the defective gene from their mother
- Fathers do not pass X-linked disorders to sons
Cause:
- Mutation in the dystrophin gene (largest known human gene)
- Dystrophin is absent or nonfunctional, leading to muscle fiber breakdown
Key Features:
- Onset: Around age 5
- Progressive muscle weakness (starts in pelvic girdle muscles)
- Gower’s sign: Child uses hands to push off thighs to stand up
- Calf muscle pseudohypertrophy:
- Calf appears large due to fat and fibrous tissue, not real muscle growth
- Thigh muscles are weak and wasted
Diagnosis:
- Elevated creatine kinase (CK)
- Genetic testing for dystrophin gene mutations
- Muscle biopsy (less common today)
Prognosis:
- Respiratory failure
- Poor
- Most patients are wheelchair-bound by teenage years
- Common cause of death:
- Dilated cardiomyopathy
2. Becker Muscular Dystrophy (BMD)
Genetics:
- X-linked recessive (same gene as Duchenne: dystrophin)
- But here, dystrophin is partially functional, not completely absent
- Caused by a partial dystrophin defect (some functional dystrophin is present)
Onset:
- Later onset, usually in adolescence or early adulthood
Symptoms:
- Milder muscle weakness than Duchenne
- Gower’s sign may be present but appears later
- Calf muscle pseudohypertrophy
- Slower progression
Diagnosis:
- Elevated CK (Creatine Kinase)
- Genetic testing (shows dystrophin mutation, but less severe than Duchenne)
Prognosis:
- Better than Duchenne
- Many patients remain ambulatory into adulthood
- Life expectancy is longer, though cardiac issues (like dilated cardiomyopathy) may still occur
3. Myotonic Dystrophy
- Inheritance: Autosomal dominant
- Typical Age of Onset: 20 to 30 years
- Genetic Cause: CTG trinucleotide repeat expansion
Clinical Features
- Gonadal atrophy (testicular atrophy or infertility)
- Myotonia: Delayed muscle relaxation; for example, difficulty releasing a handshake
- Cataracts
- Frontal balding
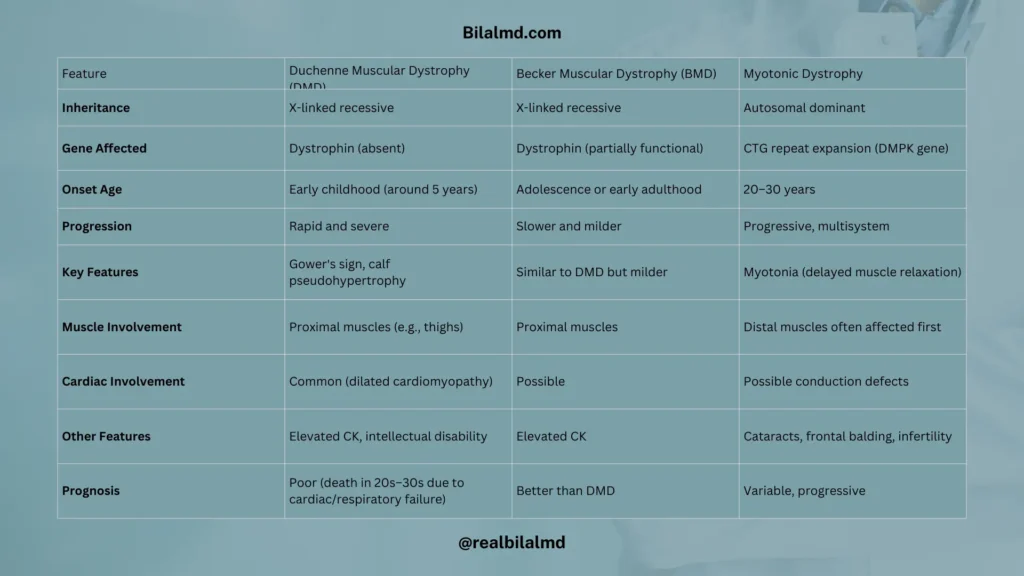
Comparison of DMD, BMD, and Myotonic Dystrophy
| Feature | Duchenne Muscular Dystrophy (DMD) | Becker Muscular Dystrophy (BMD) | Myotonic Dystrophy |
|---|---|---|---|
| Inheritance | X-linked recessive | X-linked recessive | Autosomal dominant |
| Gene Affected | Dystrophin (absent) | Dystrophin (partially functional) | CTG repeat expansion (DMPK gene) |
| Onset Age | Early childhood (around 5 years) | Adolescence or early adulthood | 20–30 years |
| Progression | Rapid and severe | Slower and milder | Progressive, multisystem |
| Key Features | Gower’s sign, calf pseudohypertrophy | Similar to DMD but milder | Myotonia (delayed muscle relaxation) |
| Muscle Involvement | Proximal muscles (e.g., thighs) | Proximal muscles | Distal muscles often affected first |
| Cardiac Involvement | Common (dilated cardiomyopathy) | Possible | Possible conduction defects |
| Other Features | Elevated CK, intellectual disability | Elevated CK | Cataracts, frontal balding, infertility |
| Prognosis | Poor (death in 20s–30s due to cardiac/respiratory failure) | Better than DMD | Variable, progressive |
5. Autosomal Trisomy
Comparison of Down Syndrome, Edwards Syndrome, and Patau Syndrome
| Feature | Down Syndrome | Edward Syndrome | Patau Syndrome |
|---|---|---|---|
| Chromosome Affected | 21 | 18 | 13 |
| Maternal Age Risk | Increases with maternal age | Increases with maternal age | Increases with maternal age |
| Facial Features | Flat face | Small chin (micrognathia) | Microcephaly |
| Hand/Foot Features | Single palmar crease, gap between 1st & 2nd toe | Rocker-bottom feet | Polydactyly |
| Gastrointestinal Issues | Duodenal atresia, Hirschsprung disease | Rare | Polycystic kidney disease |
| Neurological | Intellectual disability, early-onset Alzheimer’s | Severe intellectual disability | Holoprosencephaly |
| Other Physical Findings | Increased amyloid, leukemia (AML/ALL), hypotonia | Low-set ears | Cutis aplasia (scalp defects), |
| Prognosis | Survive into adulthood | Most die within 1 year | Most die within 1 year |
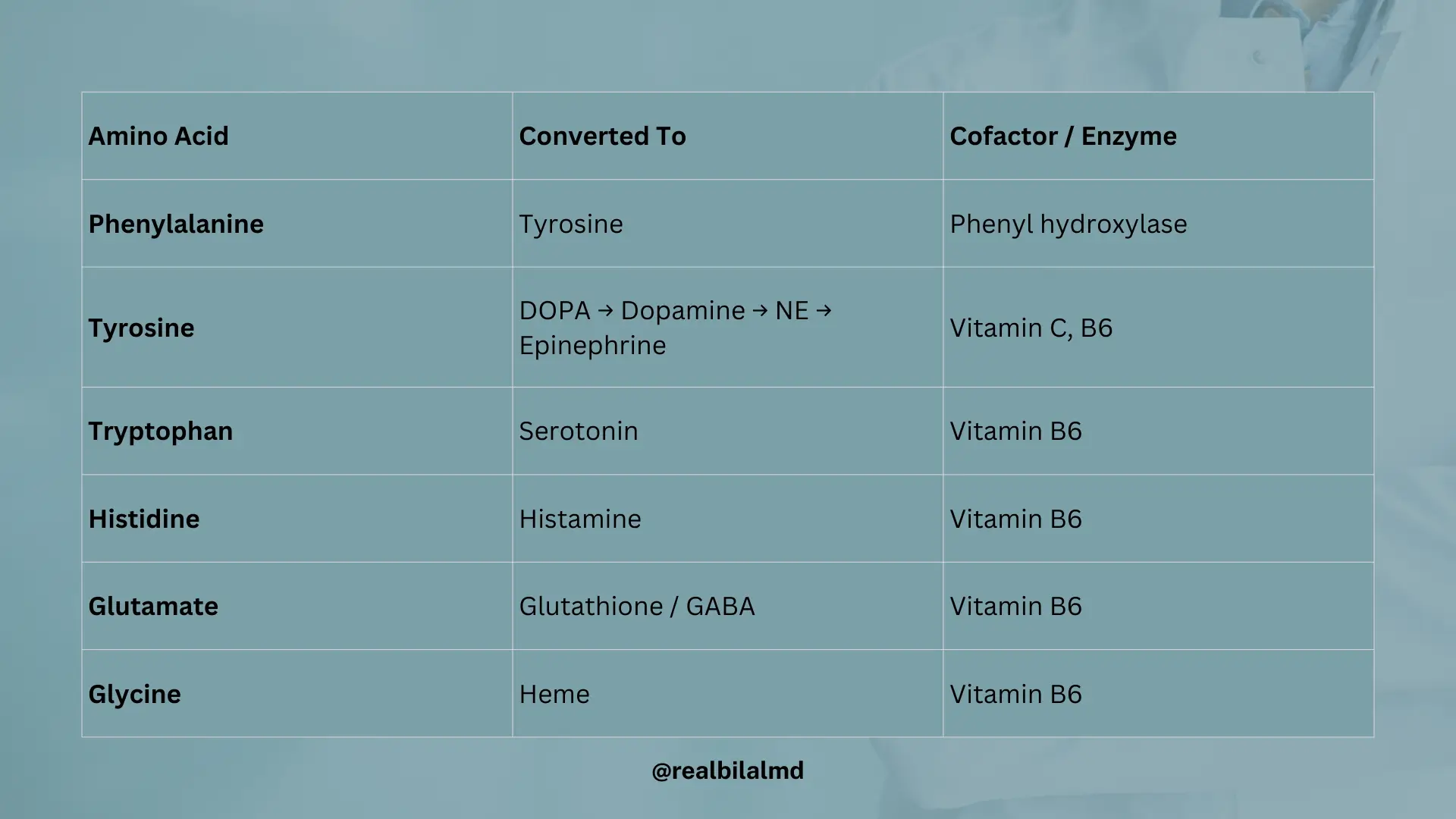
6. Amino acid derivative
| Starting Compound | Converted To | Cofactor / Enzyme |
|---|---|---|
| Phenylalanine | Tyrosine | Phenyl hydroxylase |
| Tyrosine | DOPA | |
| DOPA | NE (norepinephrine) | |
| NE | Epinephrine | |
| Tryptophan | Serotonin | Vitamin B6 |
| Tryptophan | Niacin | Vitamin B2, B6 |
| Histidine | Histamine | Vitamin B6 |
| Glutamate | Glutathione or GABA | Vitamin B6 |
| Glycine | Heme | Vitamin B6 |
7. Fructose Metabolism Disorder
Amino acids are the building blocks of proteins.
Fructose Metabolism Disorders (Autosomal Dominant)
| Step | Enzyme | Disorder | Key Features | Treatment |
|---|---|---|---|---|
| Fructose → Fructose-1-Phosphate | Fructokinase | Essential Fructosuria | AsymptomaticBenignNo treatment needed | No treatment |
| Fructose-1-Phosphate → DHAP + Glyceraldehyde | Aldolase B | Hereditary Fructose Intolerance | Hypoglycemia (neurological symptoms) Hepatomegaly, Jaundice, Nausea, vomiting | Avoid fructose and sucrose |
Galactose Metabolism Disorders
| Step | Enzyme | Disorder | Key Features | Mechanism / Notes |
|---|---|---|---|---|
| Galactose → Galactose-1-Phosphate | Galactokinase | Galactokinase Deficiency | Galactose builds up → converted to galactitol via aldose reductaseInfantile cataractsGalactose in blood | Galactose ↔ Galactitol → cataract |
| Galactose-1-P → Glucose-1-P | UDT (Uridyltransferase) | Classic Galactosemia | HepatomegalyHypoglycemiaJaundiceNausea, vomiting | Galactose-1-P → back-converted to galactose → cataract |
8. Phenylalanine
| Pathway / Enzyme | Deficiency / Disease | Key Features |
| Phenylalanine → Tyrosine (Phenylalanine hydroxylase) | Phenylketonuria (PKU) | microcephaly, intellactual disability, musty ordr urine smell, seizure |
| Tyrosine → DOPA → Dopamine → NE (vitamin C) → Epinephrine (vitamin B6) | – | normal pathway, no disorder info given |
| DOPA → Melanin (Tyrosinase) | Albinism | tyrosine decreases, ↓ melanin |
| Homogentisic acid → Fumarate (Homogentisate oxidase) | Alkaptonuria | black urine, homogentisic acid deposit on various area, anthalgia |
| Branched amino acids (Isoleucine, Leucine, Valine) → α-ketoacid dehydrogenase | Maple Syrup Urine Disease | vomiting, poor feeding, urinesmell like burnt sugar, progressive neurological symptoms |
9. Lipoproteins (Lipids and Proteins)
Lipid Transport Summary Table
| Lipoprotein | Function | Source | Destination | Main Content |
|---|---|---|---|---|
| Chylomicron | Carries exogenous triglycerides (TG) from intestine to tissues | Intestine | Peripheral tissues | Triglycerides |
| VLDL | Carries endogenous triglycerides (TG) from liver to tissues | Liver | Peripheral tissues | Triglycerides |
| LDL | Transports cholesterol from liver to peripheral tissues | From VLDL remnant | Peripheral tissues | Cholesterol |
| HDL | Carries cholesterol from peripheral tissues back to liver (good) | Peripheral tissues | Liver | Cholesterol, Proteins |
Lipoprotein Size-Density-Protein Relationship
- Size ∝ 1 / Density
(As density increases, size decreases) - Density ∝ Protein Content
(As protein content increases, density increases)
Implications:
- HDL → High protein → High density → Small size
- Chylomicron → Low protein → Low density → Large size
9. Ketone Bodies
Ketone bodies are substances that make the blood acidic, especially during prolonged fasting or uncontrolled diabetes. They serve as the last source of energy for the brain when glucose is not available. The primary ketone body is acetoacetate, while β-hydroxybutyrate and acetone are considered secondary ketone bodies. These compounds are produced in the liver and their accumulation leads to metabolic acidosis due to increased acidity in the body.

Synthesis Pathway
- Rate-limiting enzyme:
- HMG-CoA Synthase
- In Type 1 Diabetes Mellitus (DM1):
- No insulin → ↑ lipolysis → ↑ free fatty acids → ↑ Acetyl-CoA
- Acetyl-CoA condenses → Ketone body production ↑
- [H⁺] increases → blood pH decreases → Metabolic acidosis
Anion Gap Calculation
Anion Gap Formula
Standard Formula:
Anion Gap=(Na++K+)−(Cl−+HCO3−)
Alternate (Most Common) Formula:
Anion Gap=Na+−(Cl−+HCO3−)
✅ Normal Range (without K⁺): 12 – 14
✅ Normal Range (with K⁺): 14 – 18
Note: In diabetic ketoacidosis (DKA), anion gap is typically increased due to accumulation of ketone bodies (unmeasured anions).
10. Marfan Syndrome
- Type: Connective tissue disorder
- Chromosomal Involvement: Chromosome 15
- Inheritance: Autosomal Dominant (50%)
- Gene Affected: Fibrillin-1
- Body Features:
- 1. Tall stature
- 2. Thin limbs
- 3. Hypermobile joints
11. Fragile X Syndrome
- Inheritance: X-linked dominant
- Genetic Mechanism:
- Trinucleotide Repeat: CGG
- Premutation: 50–200 repeats
- Full Mutation: >200 repeats
- Key Features:
- Chin protrusion
- Gonadal dysfunction (macrogonadism)
- Long face
- Mental retardation
12. Trinucleotide Repeat Expansion Disorders
| Trinucleotide Repeat | Disease | Inheritance Pattern |
|---|---|---|
| CGG | Fragile X Syndrome | X-linked Dominant |
| CAG | Huntington Disease | Autosomal Dominant |
| CTG | Myotonic Dystrophy | Autosomal Dominant |
| GAA | Friedreich Ataxia | Autosomal Recessive |
13. Amino Acid Essential
Essential Amino Acids (Mnemonic: PVT TIM HALL)
- P – Phenylalanine
- V – Valine
- T – Threonine
- T – Tryptophan
- I – Isoleucine
- M – Methionine
- H – Histidine
- A – Arginine
- L – Leucine
- L – Lysine
Non-Essential Amino Acids
- Tyrosine: Non-essential because it can be synthesized from phenylalanine.
Ketogenic vs. Glucogenic Amino Acids
- Ketogenic: Amino acids that can be converted into ketone bodies.
- Leucine
- Lysine
- Glucogenic: Amino acids that can be converted into glucose.
- Methionine
- Histidine
- Valine
Basic Amino Acids
- Histidine (strongest base)
- Arginine
- Lysine
CPS1 and Urea Cycle
- CPS1 (Carbamoyl Phosphate Synthetase I): Enzyme that plays a key role in the urea cycle by converting ammonia (NH₃) and CO₂ into carbamoyl phosphate, which then enters the cycle.
- Defects in CPS1 result in hyperammonemia, a condition characterized by elevated ammonia levels in the blood, leading to toxic effects.
Symptoms of Hyperammonemia
- Flapping tremor (also called asterixis)
- Slurring of speech
- Vomiting
- Cerebral edema
- Lactic acidosis due to the accumulation of ammonia
- Decreased ATP due to a reduction in the citric acid cycle (alpha-ketoglutarate levels decrease)
Treatment of Hyperammonemia
- Lactulose: A laxative that helps lower ammonia levels by acidifying the gut and promoting the excretion of ammonia.
- Rifaximin: An antibiotic that reduces the intestinal production of ammonia by gut bacteria.
Check your NRE Step 1 result after completing the exam.





